Stabsstelle Kommunikation und Marketing
Forschung zu seltenen Stoffwechselkrankheiten
„HMGCLD“ ist die Kurzform für den sperrigen Namen „3-hydroxy-3-methylglutaryl-coenzyme A lyase deficiency“, eine Stoffwechselkrankheit, bei der die Aminosäure Leuzin nicht richtig abgebaut wird und Ketonkörper nicht im üblichen Maß gebildet werden können. Ketonkörper sind vor allem als Energieträger für den Körper bekannt und können in Hungerphasen einer Unterzuckerung entgegenwirken. Bei „MATD“, der „2-methylacetoacetyl-coenzyme A thiolase deficiency“, einer anderen Krankheit, können Ketonkörper zwar gebildet werden, doch können sich diese Säuren anstauen, weil ihre Verwertung gestört ist.
Beide Krankheiten werden vererbt und gehören als seltene angeborene Stoffwechselstörungen zum Forschungsgebiet von Professor Sass vom Fachbereich Angewandte Naturwissenschaften und vom Institut für Funktionale Gen-Analytik (IFGA) der H-BRS. Im Förderprogramm „Zeit für Forschung“ des Wissenschaftsministeriums des Landes NRW widmet sich Sass drei Jahre lang intensiv diesem Thema und legt nun im letzten Jahr der Förderung zwei weitere Publikationen vor.
„Seltene Stoffwechselstörung“ meint wirklich selten. Sass hat zusammen mit Privatdozentin Dr. Sarah Grünert vom Pädiatrischen Stoffwechselzentrum der Uniklinik Freiburg für die Studie weltweit gesucht und nicht mehr als 211 (HMGCLD) bzw. 244 (MATD) in der Fachliteratur dokumentierte Patienten gefunden. Laborchemisch zeichnen sich beide Krankheiten durch charakteristische Muster der organischen Säuren im Urin aus. Eine Bestätigungsuntersuchung kann durch spezifische Enzymaktivitätstests und Genanalysen erfolgen. Zwei Fünftel der Patienten mit HMGCLD wurden schon als Neugeborene symptomatisch, 80 Prozent im ersten Lebensjahr. Dies unterstreicht eine besondere Bedeutung der Ketonkörper-Bildung schon bald nach der Geburt. Trotz oft früher und schwerer Stoffwechselkrisen ließ sich für die Mehrzahl der HMGCLD-Patienten eine normale Entwicklung zeigen.
Beim MATD-Mangel gab es bei 90 Prozent der bekannten Patienten mindestens eine Stoffwechselentgleisung. Zur ersten Krise kam es meist in den ersten beiden Lebensjahren, aber kaum in der Neugeborenenzeit und nicht mehr nach dem achten Geburtstag. In Einzelfällen präsentierten sich die Betroffenen nicht mit akuten Entgleisungen, sondern mit chronischen neurologischen Symptomen. Bei 75 Prozent der beschriebenen Patienten war die Entwicklung unbeeinträchtigt und ohne neurologische Auffälligkeiten. Es scheint besonders wichtig zu sein, dass die erste Entgleisung des Stoffwechsels früh erkannt und angemessen behandelt wird.
Publikationen
Die Publikationen zu HMGCLD und MATTD sind gerade in der Fachzeitschrift „Orphanet Journal of Rare Diseases“ erschienen und kostenfrei zugänglich unter
https://ojrd.biomedcentral.com/track/pdf/10.1186/s13023-020-1319-7
und https://ojrd.biomedcentral.com/track/pdf/10.1186/s13023-020-01357-0.
Hintergrund
Die Ketonkörper Acetessigsäure und (R)-3-Hydroxy-n-Buttersäure sind Substanzen, die vor allem im Hungerzustand zur Energieversorgung des Gehirns beitragen. In den vergangenen Jahren wurde immer deutlicher, dass Ketonkörper neben ihrer Rolle als Energieträger weitere biologische Funktionen übernehmen können. Selbst bei Krebserkrankungen und Alterungsprozessen können Ketonkörper eine Rolle spielen.
Professor Dr. Jörn Oliver Sass erforscht seit vielen Jahren, vor allem mittels biochemischer und molekularbiologischer Verfahren seltene erbliche Stoffwechselstörungen. Ihm ist es zu verdanken, dass die H-BRS als einzige Fachhochschule in das „European registry and network for Intoxication type Metabolic Diseases“ (E-IMD) aufgenommen wurde, dem sonst fast ausschließlich Universitätskliniken angehören.
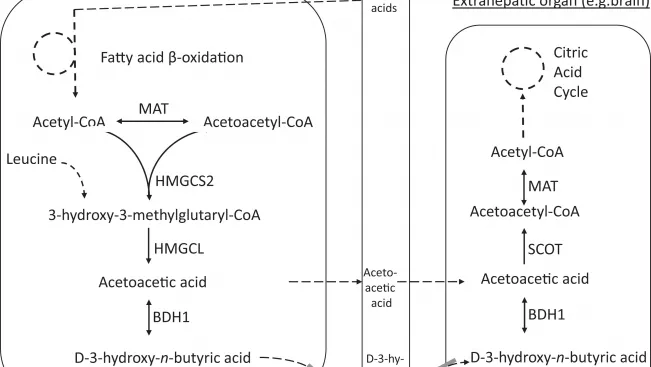
Nähere Beschreibung aus der Publikation zu obiger Abbildung:
Ketonkörperstoffwechsel und -transport
Schematische Darstellung der Enzyme und Transporter des Ketonkörperstoffwechsels, die eindeutig mit angeborenen Fehlern in Zusammenhang stehen. Sie ist nicht erschöpfend. Insbesondere können zusätzliche Monocarboxylat-Transporter (MCT) am Ketonkörper-Transfer beteiligt sein, und über den Transport von Acetessigsäure liegen nur sehr begrenzte Informationen vor. Auch andere Stellen als die Leber können kleine Beiträge zur Ketogenese liefern, z. B. Astrozyten und neugeborener Darm. Mitochondriale Acetoacetyl-CoA-Thiolase (MAT) und D-3-Hydroxy-n-Butyrat-Dehydrogenase (BDH1) sind sowohl an der Ketogenese als auch an der Ketolyse beteiligt. In den Lebermitochondrien, während der Ketogenese, gleichen MAT und andere Thiolasen die Pools von Acetoacetyl-CoA (AcAc-CoA) und Acetyl-CoA (Ac-CoA) aus. Im Gegensatz dazu ist in extrahepatischen Geweben während der Ketolyse die MAT die wichtigste mitochondriale Thiolase, die diese Reaktion ausführt, und die kritischste physiologische Rolle der MAT ist die der Ketonkörperverwertung. Die angebliche Rolle von MCT7 (kodiert durch SLC16A6) ist bisher nur im Zebrafisch bestätigt wurde.
Sie haben noch Fragen?

Jörn Oliver Sass
Professor für Bioanalytik und Biochemie, Forschungsgruppe Angeborene Stoffwechselstörungen (FG Sass)
Standort
Rheinbach
Raum
I 218; Labor F 012
Adresse
von-Liebig-Str. 20
53359 Rheinbach
Telefon
+49 2241 865 9668Links
Weiterführende Links